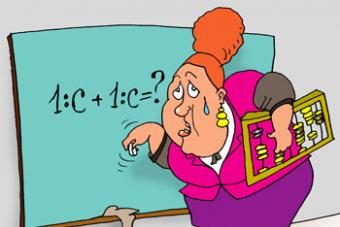Синдром Альпорта - генетически детерминированное воспалительное заболевание почек, сопровождающееся поражением слухового и зрительного анализаторов. Это достаточно редкая наследственная патология, встречающаяся у 1 из 10 тысяч новорожденных детей. По данным ВОЗ лица с синдромом Альпорта составляют 1% от всех больных с дисфункцией почек. По МКБ-10 заболевание имеет код Q87.8.
При синдроме Альпорта поражается ген, кодирующий строение белка коллагена, расположенного в базальной мембране почечных канальцев, внутреннего уха и органа зрения. Основная функция базальной мембраны – поддержание и отделение тканей друг от друга. Наследственная неиммунная гломерулопатия проявляется гематурией, нейросенсорной тугоухостью, расстройством зрения. По мере прогрессирования синдрома у больных развивается почечная недостаточность, к которой присоединяются заболевания глаз и ушей. Болезнь носит прогрессирующий характер и не поддается лечению.

Наследственный нефрит или семейный гломерулонефрит - наименования одной и той же патологии. Впервые ее описал в 1927 году ученый из Великобритании Артур Альпорт. Он наблюдал за членами одной семьи, которые страдали тугоухостью и имели эритроциты в анализах мочи. Спустя несколько лет были выявлены поражения глаз у лиц с данным заболеванием. И только в 1985 году ученые установили причину таких аномалий. Ею стала мутация гена, отвечающего за синтез и строение коллагена IV типа.
Чаще всего именно этот недуг становится причиной тяжелой почечной дисфункции у лиц мужского пола. Женщины могут передать мутантный ген своим детям, не имея клинических проявлений. Синдром манифестирует с первых лет жизни. Но чаще всего обнаруживается у малышей в возрасте 3-8 лет. У больных детей сначала появляются признаки поражения почек. Проблемы со слухом и зрением развиваются несколько позже. В позднем детском и юношеском возрасте формируется тяжелая патология почек, потеря зрения и слуха.
По способу наследования аномалии выделяют 3 формы патологии: Х-сцепленная доминантная, аутосомно-рецессивная, аутосомно-доминантная. Каждой форме соответствуют те или иные морфологические и функциональные изменения внутренних органов. В первом случае развивается классическая форма, при которой воспаление почечной ткани проявляется кровью в моче и сопровождается снижение слуха и зрения. При этом болезнь имеет прогрессивное течение, быстро развивается недостаточность почек. Гистологической особенностью подобных процессов является истончение базальной мембраны. Во втором случае врожденный недуг протекает намного легче и отличается изолированным воспалением почек с гематурией. Аутосомно-доминантная форма также считается доброкачественной, отличается благоприятным прогнозом и проявляется только гематурией или же протекает бессимптомно.

Обнаруживают наследственное воспаление почек случайно, во время профосмотра или диагностического обследования других заболеваний.
Этиология
Истинные этиопатогенетические факторы патологии до сих пор полностью не изучены. Предполагают, что синдром Альпорта - наследственное заболевание, обусловленное мутацией гена, расположенного в длинном плече Х хромосомы и кодирующего белок коллаген IV типа. Основная функция коллагена – обеспечение прочности и эластичности соединительнотканных волокон. При данном синдроме отмечается поражение сосудистой стенки почек, кортиева органа, капсулы хрусталика.
Мутантный ген чаще всего передается от родителей детям. Существует основные формы наследования патологии:

- Доминантный Х-сцепленный тип наследования характеризуется передачей пораженного гена от матери сыну или дочери, а от отца – только дочери. Синдром более тяжело протекает у мальчиков. У больных отцов рождаются здоровые сыновья и больные дочери.
- Аутосомно-рецессивный тип характеризуется получением одного гена от отца, а второго - от матери. Больные дети рождаются в 25% случаев, причем одинаково часто как среди девочек, так и среди мальчиков.
В семье с наследственными заболеваниями мочевыделительной системы вероятность рождения больных детей увеличивается в разы. Если больной ребенок рождается в семье, где все члены имеют идеально здоровые почки, причиной синдрома является спонтанная генетическая мутация.
Факторы, способствующие развитию болезни:
- родственники с почечными патологиями;
- родственные браки;
- сдвиги со стороны иммунной системы;
- снижение слуха в молодом возрасте;
- острые инфекции бактериального или вирусного происхождения;
- вакцинация;
- физическое перенапряжение.
Экспрессия мутантного гена у разных индивидуумов варьируется от слабой до значительной выраженности клинических проявлений наследственного нефрита. Процесс разрушения базальной мембраны находится в непосредственной зависимости от тяжести патологического процесса.
Патогенез
Патогенетические звенья синдрома:
- нарушение биосинтеза коллагена или его дефицит,
- деструкция базальной мембраны почек, внутреннего уха и глазного аппарата,
- прорастание коллагеновых волокон V и VI типов,
- поражение почечных клубочков,
- иммунонегативный гломерулит,
- гиалиноз клубочков, атрофия канальцев и фиброз стромы почек,
- гломерулосклероз,
- скопление в почечной ткани липидов и липофагов,
- снижение в крови уровня Ig A, повышение IgM и G,
- снижение активности Т- и В-лимфоцитов,
- нарушение фильтрационной функции почек,
- дисфункция органа зрения и слуха,
- накопление в крови токсинов и продуктов обмена,
- протеинурия,
- гематурия,
- развитие острой почечной недостаточности,
- смерть.

Заболевание развивается постепенно с ренальных симптомов. На ранних стадиях патологии почки работают полноценно, в моче имеются следы белка, лейкоцитов и крови. Поллакиурия и никтурия сопровождаются гипертензией и другими признаками мочевого синдрома. У больных расширяются чашечки и лоханки почек, возникает аминоацидурия. Спустя некоторое время присоединяется тугоухость неврогенного происхождения.
Мужчины в наибольшей степени подвержены развитию почечной дисфункции. При отсутствии лечения смерть наступает в возрасте 15-30 лет. Женщины обычно страдают скрытой формой патологии с признаками гематурического синдрома и незначительным снижением слуха.
Симптоматика
Наследственный нефрит у детей может протекать по гломерулонефротическому или пиелонефротическому типу. Клинические признаки синдрома Альпорта условно делятся две большие группы – ренальные и экстраренальные.

Основными проявлениями почечной симптоматики являются: гематурия – кровь в моче и протеинурия – белок в моче. Эритроциты в моче у больных детей появляются сразу после рождения. Сначала это бессимптомная микрогематурия. Ближе к 5-7 годам кровь в моче становится отчетливо видна. Это патогномоничный признак синдрома Альпорта. Интенсивность гематурии возрастает после острых инфекционных заболеваний - ОРВИ, ветряной оспы, кори. Активные физические нагрузки и профилактические прививки также могут спровоцировать значительное повышение эритроцитов в крови. Несколько реже у мальчиков развивается протеинурия. У девочек этот симптом обычно отсутствует. Потеря белка с мочой сопровождается отеками, повышением артериального давления, общей интоксикацией организма. Возможна лейкоцитурия без бактериурии, анемия.
Развиваясь, болезнь Альпорта осложняется развитием почечной недостаточности. Ее классические признаки - сухая, желтоватая кожа, снижение тургора, сухость во рту, олигурия, тремор кистей, ломота в мышцах и суставах. При отсутствии правильного лечения возникает терминальная стадия патологии. В таких случаях поможет поддержать жизненные силы организма только гемодиализ. Своевременная заместительная терапия или пересадка больной почки позволяют продлить жизнь больным.
К внепочечным симптомам относятся:
- тугоухость, обусловленная невритом слухового нерва;
- ухудшение зрения, связанное с катарактой, изменением формы хрусталика, появлением белых или желтых вкраплений на сетчатке в районе макулы, миопией, кератоконусом;
- задержка в психофизическом развитии;
- врожденные дефекты – высокое небо, синдактилия, эпикант, деформация ушей, патологии прикуса;
- лейомиоматоз пищевода, трахеи, бронхов.
К неспецифическим общеинтоксикационным признакам патологии относятся:
- головная боль,
- миалгия,
- головокружение,
- резкие колебания артериального давления,
- одышка,
- частое, поверхностное дыхание,
- шум в ушах,
- бледность кожи,
- частые позывы к мочеиспусканию,
- диспепсия,
- ухудшение аппетита,
- нарушение режима сна и бодрствования,
- зуд кожи,
- судороги,
- боль в груди,
- спутанность сознания.
У больных развивается компенсированная клубочковая и канальцевая недостаточность, нарушается транспорт аминокислот и электролитов, концентрационная способность почек, ацидогенез, поражается система канальцев нефрона. По мере прогрессирования патологии признаки мочевого синдрома дополняются выраженной интоксикацией, астенизацией и анемизацией организма. Подобные процессы развиваются у мальчиков, имеющих пораженный ген. У девочек заболевание протекает намного легче, стойкая дисфункция почек у них не развивается. Только во время беременности девушки страдают от симптомов недуга.
Осложнения синдрома Альпорта развиваются при отсутствии адекватной терапии. У больных нарастают признаки недостаточности почек: появляются отеки на лице и конечностях, гипотермия, охриплость, олигурия или анурия. Часто присоединяется вторичная бактериальная инфекция – развивается пиелонефрит или гнойный отит. В таком случае прогноз неблагоприятный.
Диагностика

Диагностикой и лечением синдрома Альпорта занимаются педиатры, нефрологи, генетики, ЛОР-врачи, офтальмологи.
Диагностические мероприятия начинаются со сбора анамнеза и выслушивания жалоб больного. Особое значение имеет семейный анамнез. Специалисты выясняют, имелись ли случаи гематурии или протеинурии у родственников, а также случаи смерти от почечной дисфункции. Для постановки диагноза важны данные генеалогического анализа и акушерского анамнеза.
- Специфическое поражение базальной мембраны у больных обнаруживают по результатам биопсии.
- В общем анализе мочи - эритроциты, белок, лейкоциты.
- Генетическое исследование – выявление мутации генов.
- Аудиометрия обнаруживает нарушения слуха.
- Обследование у офтальмолога позволяет выявить врожденную патологию зрения.
- Ультразвуковое исследование почек и мочеточников, магнитно-резонансная томография, рентген и сцинтиграфия - дополнительные диагностические методики.
Лечение
Синдром Альпорта - неизлечимое заболевание. Замедлить развитие почечной недостаточности помогут следующие рекомендации специалистов:
- Рациональное и витаминизированное питание,
- Оптимальные физические нагрузи,
- Частые и длительные прогулки на свежем воздухе,
- Санация очагов хронической инфекции,
- Профилактика инфекционных заболеваний,
- Запрет на стандартные прививки больным детям,
- Фитосбор из крапивы, тысячелистника и черноплодной рябины показан больным детям с гематурией,
- Витаминотерапия и биостимуляторы для улучшения обмена веществ.
Правильное питание заключается в употреблении легкоусвояемых продуктов с достаточным содержанием основных нутриентов. Из рациона больных следует исключить солености и копчености, пряные и острые блюда, алкоголь, продукты с искусственными красителями, стабилизаторами, ароматизаторами. В случае нарушения функций почек необходимо ограничить потребление фосфора и кальция. Подобные рекомендации следует соблюдать больным в течение всей жизни.

Медикаментозная симптоматическая терапия:
- Для устранения гипертензии назначают ингибиторы АПФ – «Каптоприл», «Лизиноприл» и блокаторы рецепторов ангиотензина – «Лориста», «Вазотенз».
- Пиелонефрит развивается в результате присоединения инфекции. В таком случае применяют антибактериальные и противовоспалительные медикаменты.
- Для коррекции нарушений водно-электролитного обмена назначают «Фуросемид», «Верошпирон», внутривенно физраствор, глюкозу, кальция глюконат.
- Анаболические гормоны и железосодержащие препараты показаны для ускоренного образования эритроцитов.
- Иммуномодулирующая терапия – «Левамизол».
- Антигистаминные препараты – «Зиртек», «Цетрин», «Супрастин».
- Комплекс витаминов и лекарств, улучшающих обмен веществ.
Гипербарическая оксигенация оказывает положительный эффект на выраженность гематурии и функционирование почек. При переходе почечной недостаточности в терминальную стадию требуется гемодиализ и пересадка почки. Оперативное вмешательство проводится после достижения больными пятнадцатилетнего возраста. Рецидив заболевания в трансплантате не отмечается. В отдельных случаях возможно развитие нефрита.
Генотерапия синдрома в настоящее время активно разрабатывается. Ее основная цель – предупреждение и замедление ухудшения функционирования почек. Этот перспективный вариант лечения сегодня внедряется в лечебную практику западными медицинскими лабораториями.
Прогноз и профилактика
Синдром Альпорта - наследственное заболевание, предупредить появление которого просто невозможно. Соблюдение всех предписаний врача и ведение здорового образа жизни помогут улучшить общее состояние больных.
Прогноз синдрома считается благоприятным, если у больных обнаруживается гематурия без протеинурии и тугоухости. Почечная недостаточность не развивается также у женщин без поражения слухового анализатора. Даже при наличии стойкой микрогематурии заболевание у них практически не прогрессирует и не ухудшает общего состояния больных.
Наследственный нефрит в сочетании с быстрым развитием почечной недостаточности имеет неблагоприятный прогноз у мальчиков. У них рано развиваются дисфункции почек, глаз и ушей. При отсутствии своевременного и грамотного лечения больные погибают в возрасте 20-30 лет.
Синдром Альпорта – опасное заболевание, которое без оказания квалифицированной медицинской помощи ухудшает качество жизни пациентов и заканчивается их смертью. Чтобы облегчить течение наследственного нефрита, необходимо неукоснительно соблюдать все врачебные рекомендации.
Видео: лекция по синдрому Альпорта
В последние несколько столетий широко развивается научно-технический прогресс, создаются массивные фабрики, заводы и предприятия. Выброс различных токсинов в атмосферу, водные массивы и почву часто приводит к развитию врождённых заболеваний. Одним из редких недугов такого плана является синдром Альпорта - патология, поражающая сразу несколько систем человеческого тела (в особенности страдают почки и органы чувств). Бороться с этой формой заболевания довольно трудно, а также существует высокая вероятность передачи её последующим поколениям.
Определение синдрома Альпорта
Синдром Альпорта - это врождённое заболевание, связанное с возникновением в организме патологический мутаций. В результате этого происходит образование неправильного коллагена, нарушающего функционирование органов или тканей. Источником мутировавшего гена может быть один из родителей ребёнка (чаще недуг передаётся от матери). Патология встречается крайне редко (менее 15 человек на 100.000 населения).
Триада, типичная для синдрома Альпорта, обнаруживается в 50% случаев
Так как развитию заболевания подвержены в основном люди одной родственной группы, его можно называть наследственным нефритом. Считается, что вероятность формирования недуга увеличивается при кровосмешении.
Какие разновидности патологии существуют:
- изолированное поражение почечных клубочков;
- наличие тугоухости или глухоты в сочетании с повреждением почек и нарушениями зрения;
- глухота, миопия, нефрит и внешние уродства;
- бессимптомное носительство.
Причины развития недуга
В основе формирования всех симптоматических проявлений недуга лежит генная мутация. В организме человека на молекулярном уровне реализуется сбой, в результате которого нарушается нормальная сборка и транспортировка коллагена - белка, входящего в состав практически всех органов и тканей человеческого тела. Заболевание носит наследственный характер и может передаваться пациенту от его предков следующими путями:
- аутосомно-доминантным (встречается у человека, даже если только 1 из его родителей страдал от недуга);
- аутосомно-рецессивный (возникает у ребёнка, когда мать и отец имеют одинаковые генетические мутации);
- сцепленный с полом (женщина является бессимптомным носителем, а у мужчины формируется вся клиническая картина болезни).
Обострение и манифестация синдрома Альпорта возникают при воздействии следующих факторов:
- перенесённые ОРВИ, грипп, простуда;
- оперативное вмешательство;
- травматическое повреждение различных органов и тканей;
- стресс и нервно-психическое напряжение;
- вакцинация от инфекций;
- беременность, роды, становление менструальной функции;
- употребление наркотических препаратов, алкоголя и никотина;
- бесконтрольный приём лекарственных средств.
Клинические проявления синдрома Альпорта
Симптомы подобного заболевания у 80% пациентов появляются в первые 10 лет жизни. При носительстве мутантных генов люди могут даже не догадываться о наличии у них такого недуга, так как его типичная картина полностью отсутствует. Из-за разных клинических форм синдрома Альпорта невозможно чётко выделить ведущий признак заболевания, наблюдается сразу несколько симптомов:
- Снижение зрения. Пациент отмечает, что при недостаточном освещении пропадает чёткость букв, изображение начинает расплываться и теряться. Это напрямую связано с изменением волокон коллагена, входящих в состав хрусталика глаза. Первично удаётся скорректировать эту проблему с помощью очков и линз, но впоследствии больному может понадобиться операция.
- Ухудшение слуха или наступление полной глухоты. Чаще всего появление этого симптома замечают друзья и родственники: пациент постоянно прибавляет звук, не может воспринимать шёпотную речь, сам начинает говорить громче. Поражаются оба уха, в результате чего больной не может воспринимать низкие и высокие частоты. Пациенты также могут предъявлять жалобы на нарастающий шум внутри головы, который мешает сосредоточиться.
- Появление в моче инородных примесей. При поражении почек во время синдрома Альпорта развивается повреждение сосудов, в результате чего в урине образуется кровь. Сначала обнаруживаются единичные эритроциты, при прогрессировании болезни моча окрашивается полностью, появляются массивные сгустки. При неблагоприятном течении недуга происходит закупорка мочеточника или лоханки, в результате чего нарушается прохождение урины.
- Болезненность в поясничной области. Неприятные давящие и тянущие ощущения усиливаются при физических нагрузках, стрессах, воспалительных заболеваниях, приёме некоторых медикаментов. Их возникновение связано с деформацией почечного вещества из-за отёков.
- Перепады артериального давления. Пациенты часто предъявляют жалобы на головные боли опоясывающего характера, которые возникают после употребления солёной, жирной пищи или алкоголя. Такой симптом служит признаком развития артериальной гипертензии, сопровождающей повреждение почек. А также для больных с синдромом Альпорта типичен коллапс - расслабление периферических сосудов, в результате которого резко падает давление и пострадавший теряет сознание.
Различные методики подтверждения диагноза
Пациент с прогрессирующим снижением слуха, зрения и проблемами с мочевыделительной системой чаще всего обращается к терапевту. При наличии всех трёх основных признаков недуга сразу поставить диагноз не составляет большого труда. Однако при отсутствии одного или нескольких из них определиться гораздо тяжелее. Синдром Альпорта дифференцируют с:
- нейросенсорной тугоухостью;
- лабиринтной глухотой;
- миопией;
- катарактой;
- пиелонефритом;
- гломерулонефритом;
- злокачественной или доброкачественной опухолью почек.
Для подтверждения диагноза используют следующие методики:
- общий анализ мочи - появление белка, эритроцитов и изменение мутности урины позволяют заподозрить проблемы с почками;
- ультразвуковая диагностика выявляет патологический отёк клубочков и нарушение их структуры;
- прогрессирующее снижение частоты слуха фиксируется с помощью тимпанометрии (определяется эластичность барабанной перепонки);
- миопию выявляет офтальмолог с использованием специальных таблиц и аппаратом (снижение остроты зрения - явный симптом заболевания);
- молекулярно-генетическое типирование биологического материала (обнаруживаются мутации в геноме человека, отвечающие за образование патологического коллагена).
Синдром Альпорта - это довольно редкое заболевание, которое трудно заподозрить молодому врачу. Один из моих пациентов перед постановкой диагноза прошёл длительный путь по самым разным инстанциям. В возрасте 6 лет у него обнаружили прогрессирующее снижение слуха, после чего ребёнка отправили к ЛОР-врачу. Он подтвердил диагноз и выписал слуховой аппарат для постоянного ношения. Во время проверки зрения перед поступлением в 1 класс у малыша обнаружили сильную близорукость, которую нельзя было скорректировать с помощью очков. В подростковом возрасте мальчик начал часто болеть и постоянно испытывал проблемы с почками, что доктора ошибочно приняли за проявления пиелонефрита после гриппа или ангины. В течение нескольких месяцев пациента лечили с применением антибиотиков, а его состояние неуклонно ухудшалось. Только после этого медики решили провести генетическое исследование, в ходе которого обнаружилось наличие мутации белка коллагена. Мальчику подобрали подходящий курс терапии, после чего ему стало лучше и появилась возможность провести операцию по пересадке почки.
Каким образом проводится лечение синдрома Альпорта
Тактика после подтверждения диагноза заболевания напрямую зависит от тяжести состояния пациента. Если у него отсутствуют видимые клинические проявления почечной недостаточности и интоксикации организма, лечение проводится в домашних условиях или в дневном стационаре, куда человек должен регулярно приходить за фармацевтическими препаратами. При более тяжёлом течении недуга показана госпитализация в отделение нефрологии или урологии. Медикаментозная терапия направлена на устранение основных симптомов синдрома Альпорта, а также на защиту от повреждения почечной ткани. При отсутствии эффекта от консервативного лечения назначается гемодиализ, а после него - операция по трансплантации почек. Пациенту всё время необходимо соблюдать палатный режим и придерживаться диеты.
Основные цели терапии синдрома Альпорта:
- профилактика прогрессирования почечной недостаточности;
- нормализация водно-солевого баланса;
- защита от присоединения микробной инфекции;
- укрепление иммунитета;
- выведение из организмов продуктов распада вредных токсинов.
Таблица: применяемые медикаментозные препараты
| Название группы лекарственных средств | Основные действующие вещества | Эффекты от использования |
| Стероидные противовоспалительные медикаменты |
| Уменьшают выраженность неприятных болезненных ощущений в нижней части спины и поясничной области, снимают отёчность почечных тканей |
| Нестероидные противовоспалительные препараты |
|
|
| Иммуностимуляторы |
| Способствуют образованию защитных клеток в костном мозге, которые обеспечивают функционирование иммунитета |
| Диуретики |
| Выводят из организма лишнюю жидкость и токсичные вещества, накапливающиеся при недостаточной работе почек, нормализуют артериальное давление |
| Витаминные и минеральные комплексы |
| Восстанавливают баланс необходимых микроэлементов, которые участвуют в обменных процессах |
Фотогалерея: средства для медикаментозной терапии недуга
 Фуросемид удаляет из организма лишнюю жидкость
Фуросемид удаляет из организма лишнюю жидкость  Нимесулид обладает обезболивающим эффектом
Нимесулид обладает обезболивающим эффектом  Полиоксидоний укрепляет иммунитет
Полиоксидоний укрепляет иммунитет
Пациентом с заболеваниями почек необходимо постоянно соблюдать диету. При синдроме Альпорта нарушается нормальное усвоение питательных веществ, а также из организма не выводятся различные токсины. Для борьбы с симптомами недуга тратится огромное количество энергии, которую нужно восстановить с помощью пищи. Баланс белков, жиров и углеводов должен составлять 4:1:1. Необходимо добавлять в рацион больше витаминов группы B, C и A, а также микроэлементы натрий, калий, кальций и магний. Рекомендуется готовить только из свежих продуктов, приобретённых на фермерском рынке или у частных продавцов, чтобы избежать консервантов и вредных добавок.
Пациентам с синдромом Альпорта необходимо регулярно следить за количеством потребляемой воды. Более 2 литров в течение суток создают лишнюю нагрузку на почки.
Примерное меню питания для больных:
- Завтрак. Омлет с козьим сыром и зеленью, несколько тостов с маслом и ветчиной. Кофе рекомендуется заменить на красный чай, чтобы не провоцировать увеличение артериального давления. В качестве альтернативы омлету могут выступать различные каши на молоке или творог с добавлением фруктов.
- Обед. Суп, сваренный на нежирной говядине, курице или индейке (борщ, гороховый, щи, рассольник, харчо), а также салат из морской или квашеной капусты. При необходимости можно съесть несколько кусочков чёрного хлеба с чесноком или луком для укрепления иммунитета.
- Ужин. Постные котлеты, отварная курица или рыба с минимальным количеством соли. На гарнир можно использовать макароны твёрдых сортов, тушёные овощи (рекомендуется ограничить употребление картофеля), рис, гречку, чечевицу, нут, бобы.
Фотогалерея: полезная еда
 Творог - лучший источник кальция
Творог - лучший источник кальция  Овощи и фрукты богаты витаминами
Овощи и фрукты богаты витаминами  Крупы - источник медленных углеводов
Крупы - источник медленных углеводов
Физиотерапевтические методики для борьбы с симптомами недуга
На восстановительном этапе необходимо укрепить организм пациента и защитить его от развития вторичных осложнений. С этими задачами прекрасно справляется процедуры физиотерапии, которые основаны на применении физических явлений. Курс лечения подбирает врач-нефролог на основании данных о течении недуга. При обострении синдрома Альпорта рекомендуется отменить некоторые процедуры, так как они нанесут организму больше вреда, чем пользы.
Физиотерапевтические методики для лечения патологии:
- УВЧ-терапия основана на применении электрических полей высокой частоты. Этот способ помогает избавиться от неприятных ощущений в поясничной области, а также расслабляет спазмированные мышцы.
- Массаж назначается при отсутствии данных об опущении почек. Точечное воздействие на мягкие ткани улучшает кровообращение и отток лимфы, что защищает организм от присоединения нежелательных осложнений в виде венозного застоя.
- Лазерная терапия. Направленные лучи уменьшают интенсивность воспаления, а также препятствуют образованию патологических разрастаний соединительной ткани (спаек).
- Применение ультразвука способствует более быстрой регенерации мягких тканей и обеспечивает заживление мелких повреждений.
Фотогалерея: физиопроцедуры для борьбы с недугом
 Использование лазера защищает от развития спаек
Использование лазера защищает от развития спаек  УВЧ-терапия эффективно справляется с симптомами недуга
УВЧ-терапия эффективно справляется с симптомами недуга  Ультразвуковая терапия стимулирует регенерацию
Ультразвуковая терапия стимулирует регенерацию
Хирургические методики устранения последствий заболевания
При синдроме Альпорта поражается ткань почек (в особенности канальцевая система). Орган теряет способность к выполнению привычных функции, в результате чего необходима его замена. Подбор донора осуществляется на основании множества критериев (ближайшие родственники не подходят из-за вероятности наличия скрытой формы заболевания) и занимает до нескольких лет. Пересадка проводится пациентам с 5–6 лет.
Операция проходит следующие этапы:
- Обработка хирургического поля. Поясничную область обкладывают стерильными салфетками и протирают спиртом и йодом.
- Доктор проводит разрез, последовательно раздвигая кожу, жировую клетчатку и мышечные пучки. Из раны извлекается повреждённая почка и её ножка, включающая сосуды, мочеточник и нервные пучки. Параллельно с этим другие медицинские работники подготавливают трансплантат.
- Новая почка устанавливается немного выше предыдущей, после чего её подшивают и фиксируют к окружающим тканям. Изменённый старый орган не удаляют, чтобы он также продолжал выполнять хотя бы часть своих функций.
- Последовательное соединение кожи, жировой клетчатки и мышечных пучков. В области раны остаётся резиновая трубка - дренаж, по которому оттекает патологическое содержимое (гной, кровь). Прооперированного больного переводят в отделение реанимации для наблюдения.
Помните, что трансплантация донорской почки не поможет вам избавиться от синдрома Альпорта. В моей практике встречался пациент, который после проведённой операции резко поменял привычный образ жизни, начал активно употреблять алкоголь и выкуривать по несколько сигарет в течение суток, перестал принимать необходимые лекарства. Это привело к резкому ухудшению его состояния, мужчину госпитализировали в отделение реанимации и интенсивной терапии, так как пересаженная почка начала отторгаться. Ценой неимоверных усилий докторам удалось оставить трансплантат и предотвратить его инфицирование. Именно поэтому даже после проведения операции необходимо следить за здоровьем и принимать лекарства.
Гемодиализ при синдроме Альпорта
При длительном нарушении функции почек в организме скапливаются токсины, которые трудно удалить без постороннего вмешательства. Чтобы защитить от повреждения головной мозг, нервную систему и другие важные органы, используется гемодиализ. Эта процедура направлена на внепочечное очищение крови от вредных примесей (химических элементов, продуктов распада вредных веществ, различных микробов) и показана всем пациентам со среднетяжелым течением синдрома Альпорта. Гемодиализ назначается в следующих ситуациях:
- угрожающее повышение уровня креатинина и мочевины;
- выраженное нарушение функции почек;
- отказ одного из органов.
Лечение заключается в механическом очищении крови
Пациент находится на кушетке полулёжа, после чего ему в локтевую вену вводят специальные катетеры и трубки. По ним кровь оттекает в аппарат, пропускающий жидкость через эластичные мембраны. Таким образом кровеносное русло очищается от токсинов и инородных примесей, так как они не проходят через поры фильтра. Повторяют такое лечение по необходимости (не реже 1 раза в 6 месяцев).
При проведении гемодиализа пациенту предоставляется справка или больничный лист на все часы, которые требует процедура. Больные практически не испытывают неприятных ощущений и могут спокойно заниматься своими делами. Пациенты моего отделения предпочитают проводить время за чтением книг, просмотром фильмов или сериалов, словесными играми. Это способствует созданию комфортной психологической обстановки.
Прогнозы и нежелательные последствия заболевания
Синдром Альпорта - это неизлечимая наследственная патология, которая передаётся следующему поколению с генами одного или обоих родителей. Полного излечения не удаётся добиться даже после операциии по трансплантации почки. Вся терапия направлена на поддержание самочувствия пациента нужном уровне и защиту от развития осложнений. Ожидаемая продолжительность жизни больных с таким диагнозом снижается на 10–15 лет. На вероятность развития нежелательных последствий оказывают влияние возраст пациента, наличие у него других острых или хронических недугов, травм и инфекций, а также образ жизни и соблюдение врачебных рекомендаций.
При выявлении заболевания у детей для каждого малыша создаётся индивидуальная программа обучения и контроля за показателями здоровья, позволяющая избежать нежелательных последствий.
Во время работы в отделении нефрологии мне довелось столкнуться с мужчиной в возрасте 65 лет, у которого диагноз синдрома Альпорта был поставлен в детстве. Больной не имел никаких характерных внешних изменений, а всего анализы были крайне хорошими. В ходе опроса выяснилось, что пациент ведёт преимущественно здоровый образ жизни, регулярно вакцинируется от гриппа, кушает еду в соответствии с диетой, занимается лёгкими физическими нагрузками, не курит и не употребляет алкогольные напитки. Такое отношение мужчины к лечению позволило ему избежать операции по пересадке почки, а также надолго отказаться от проведения процедуры гемодиализа.
Возможные осложнения и нежелательные последствия, которые могут возникнуть у больных с синдромом Альпорта:
- хроническая или острая почечная недостаточность;
- создание условий для присоединения вторичной инфекции;
- иммунодефицитные состояния;
- разрушение одной или обеих почек;
- накопление в организме токсичных веществ, которые не выводятся с мочой;
- социальная дезадаптация;
- прогрессирующее ухудшение слуха и зрения;
- повышение риска развития злокачественных новообразований;
- анемия - снижение уровня гемоглобина и эритроцитов в крови;
- воспалительные заболевания почек;
- нарушения психического и физического развития (у малышей).
Особенности клинической картины и лечения синдрома Альпорта у детей
Заподозрить наличие патологии у малыша можно сразу же после его появления на свет. Дети имеют характерные изменения и деформации лицевого черепа в виде расщепления верхней губы (заячья губа) и воронкообразного твёрдого нёба (волчья пасть), увеличенные размеры головы, а также типичное выражение лица с блуждающим взглядом. Другие мутации могут проявляться в форме нарушения слуха и зрения. Такой ребёнок не реагирует даже на громкие звуки и резкие движения окружающей среды, что обнаруживается ещё в родильном доме.
Для устранения внешних дефектов маленьким пациентам проводится пластическая операция. Мне довелось участвовать в ушивании верхней губы ребёнку с такой проблемой. После хирургического вмешательства остаётся небольшой шрам, который со временем бледнеет и становится практически незаметным. Операции проводятся в течение нескольких недель после рождения ребёнка, что снижает риск возникновения вторичных осложнений.
Для профилактики повреждений почечной ткани маленьким пациентам назначается специальная диета. Новорождённые и младенцы находятся на грудном вскармливании или получают максимально адаптированную к составу женского молока смесь. Это позволяет устранить риск развития иммунного дефицита. Основные средства для терапии симптоматических признаков заболевания вводятся на 5–7 году жизни (раньше - по потребности) и практически не отличаются от лечения, которое получает взрослый человек. Для адаптации слепых и глухих детей в обществе используют слуховые аппараты, корректирующие операции. Первые несколько лет ребёнок должен посещать специальные школы или дополнительно заниматься со специалистом, чтобы нагнать сверстников.
Фотогалерея: как выглядят дети с синдромом Альпорта
 Изменение формы черепа при заболевании встречается в 40% случаев
Изменение формы черепа при заболевании встречается в 40% случаев  Одутловатость лица - это следствие неправильной работы почек
Одутловатость лица - это следствие неправильной работы почек  Внешние пороки помогают сразу заподозрить наличие недуга
Внешние пороки помогают сразу заподозрить наличие недуга
Особенности развития болезни у девушек и влияние на репродуктивную функцию
У пациентов женского пола не всегда встречаются явные признаки синдрома Альпорта. Многие девушки узнают о носительстве подобной патологии только после рождения больного малыша. В более тяжёлых случаях заболевание активизируется в период полового созревания под воздействием гормональной перестройки организма (во время менструации) или после первого сексуального опыта.
Если состояние пациентки стабилизировано, а также не имеется других противопоказаний (поражение единственной почки, недавно перенесённая операция), можно заводить ребёнка.
В моей практике встречалась пациентка, у которой синдром Альпорта появлялся через каждые 3 поколения. У данной больной патология длительное время пребывала в спящем состоянии, но активизировалась после перенесённого стресса. Женщина отметила, что от этой формы недуга страдали также её прабабка и родная сестра. Доктора рассчитали, что вероятность появления подобной болезни у последующих поколений крайне велика.
Пациентки с тяжёлой формой синдрома Альпорта испытывают определённые трудности при зачатии и вынашивании ребёнка. Это может быть связано как с аномалиями строения репродуктивных органов, так и с нарушением работы эндокринной системы. В период роста и развития плода в материнском организме вырабатывается в 2 раза больше жидкости, в результате чего существенно возрастает нагрузка на почки. Из-за нефритических изменений воспалительного характера организм не справляется с увеличением объёма циркулирующей крови, а у женщины возникают выкидыши на различных сроках.
Какие осложнения могут возникнуть у пациенток с синдромом Альпорта при вынашивании беременности:

Наследственные заболевания почек - это группа опасных патологий которые существенно снижают не только качество жизни человека, но и её продолжительность. В настоящее время особенно актуальной является проблема ранней диагностики синдрома Альпорта. Почти все пациенты живут в неведении в течение многих лет, пока какой-либо резкий фактор не спровоцирует развитие клинических симптомов заболевания и его осложнений. Именно поэтому так важно постоянно следить за своим здоровьем, отмечая малейшие изменения в самочувствии, а также регулярно обращаться к доктору для контроля анализов.
Синдром Альпорта (СА, синоним: наследственный нефрит) – неиммунная генетически детерминированная гломерулопатия, обусловленная мутацией генов, кодирующих коллаген 4 типа базальных мембран, проявляющаяся гематурией и/или протеинурией, прогрессирующим снижением почечных функций, нередко сочетающаяся с патологией слуха и зрения (НГ).
Заболевание было впервые описано британским врачом Артуром Альпортом в 1927 году.
По эпидемиологическим данным в России частота СА среди детской популяции составляла 17:100.000 населения.
Тип наследования синдрома Альпорта может быть разным:
Х-сцепленный доминантный (XLAS): 85%.
Аутосомно-рецессивный (ARAS): 15%.
Аутосомно-доминантный (ADAS): 1%. Наиболее распространенная Х-сцепленная форма синдрома Альпорта приводит к терминальной стадии почечной недостаточности у мужчин. Гематурия обычно возникает у мальчиков с синдромом Альпорта в первые годы жизни. Протеинурия обычно отсутствует в детстве, но это состояние часто развивается у мужчин с XLAS и у представителей обоих полов с ARAS. Потеря слуха и поражение глаз никогда не обнаруживаются при рождении – они возникают в позднем детстве или в юности, незадолго до развития почечной недостаточности. Самый распространенный тип синдрома Альпорта вызван мутациями в генах, расположенных на длинном плече Х-хромосомы, отвечающих за биосинтез коллагена. Мутации в этих генах нарушают нормальный синтез коллагена типа IV, который является очень важным структурным компонентом базальных мембран в почках, внутреннем ухе и глазах. При нарушении синтеза коллагена типа IV гломерулярные базальные мембраны в почках не способны нормально фильтровать токсичные продукты из крови, пропуская в мочу белки (протеинурия) и эритроциты (гематурия). Аномалии синтеза коллагена типа IV приводят к почечной недостаточности и отказу почек, что и является главной причиной смерти при синдроме Альпорта. Клиника :
Гематурия – это наиболее частое и раннее проявление синдрома Альпорта. Микроскопическая гематурия наблюдается у 95% женщин и практически у всех мужчин. У мальчиков гематурию обычно выявляют в первые годы жизни. Если у мальчика за первые 10 лет жизни не обнаружена гематурия, то эксперты рекомендуют считать, что у него маловероятно наличие синдрома Альпорта. Протеинурия в детстве обычно отсутствует, но иногда развивается у мальчиков с Х-сцепленным синдромом Альпорта. Протеинурия, как правило, прогрессирует. Значительная протеинурия у больных женского пола встречается нечасто. Гипертензия чаще присутствует у пациентов мужского пола с Х-сцепленным типом наследования и у больных обоих полов с аутомосно-рецессивным типом. Частота и тяжесть гипертензии повышается с возрастом и по мере прогрессирования почечной недостаточности.
Нейросенсорная тугоухость (нарушение слуха) – это характерное проявление синдрома Альпорта, которое наблюдается довольно часто, но не всегда. Есть целые семьи с синдромом Альпорта, которые страдают от тяжелой нефропатии, но имеют нормальный слух. Нарушение слуха никогда не обнаруживается при рождении. Билатеральная высокочастотная нейросенсорная тугоухость обычно проявляется в первые годы жизни или в раннем подростковом возрасте. На ранней стадии болезни нарушение слуха определяется только при аудиометрии. По мере прогрессирования, нарушение слуха распространяется на низкие частоты, включая человеческую речь. После появления тугоухости следует ожидать вовлечения почек. Ученые утверждают, что при Х-связанном синдроме Альпорта 50% мужчин страдают нейросенсорной тугоухостью к 25 годам, а к 40 годам – около 90%. Передний лентиконус (выпячивание центрального участка хрусталика глаза вперед) наблюдается у 25% пациентов с Х-сцепленным типом наследования. Лентиконуса нет при рождении, но с годами он приводит к прогрессирующему ухудшению зрения, которое заставляет больных часто менять очки. Состояние не сопровождается болью в глазах, покраснением или нарушениями цветового зрения.
Ретинопатия – это самое распространенное проявление синдрома Альпорта со стороны органа зрения, поражает 85% мужчин с Х-сцепленной формой болезни. Появление ретинопатии обычно предшествует почечной недостаточности.
Диффузный лейомиоматоз пищевода и бронхиального дерева – еще одно редкое состояние, которое наблюдается в некоторых семьях с синдромом Альпорта. Симптомы появляются в позднем детском возрасте и включают нарушение глотания (дисфагия), рвоту, боль в эпигастрии и за грудиной, частые бронхиты, одышку, кашель.
На аутосомно-рецессивную форму приходится всего 10-15% случаев болезни. Эта форма встречается у детей, чьи родители являются носителями одного из пораженных генов, сочетание которых у ребенка вызывает болезнь. Сами родители не имеют симптомов или имеют незначительные проявления, а дети тяжело больны – их симптомы напоминают таковые при Х-сцепленном типом наследования.
Аутосомно-доминантная форма синдрома Альпорта – это самая редкая форма синдрома, которая затрагивает одно поколение за другим, причем мужчины и женщины болеют одинаково тяжело. Почечные проявления и глухота напоминают Х-сцепленную форму, но почечная недостаточность может возникать в более позднем возрасте. Клинические проявления аутосомно-доминантной формы дополняются склонностью к кровотечениям, макротромбоцитопенией, синдромом Эпштейна, наличием нейтрофильных включений в крови. Диагностика синдрома Альпорта
Лабораторные анализы. Анализ мочи: у больных с синдромом Альпорта чаще всего присутствует кровь в моче (гематурия), а также высокое содержание белка (протеинурия). Анализы крови демонстрирует почечную недостаточность (повышение количества лейкоцитов, повышение скорости оседания эритроцитов (СОЭ) – признак инфекции, воспалительного процесса; снижение количества эритроцитов и гемоглобина (анемия); снижение количества тромбоцитов (обычно небольшое).
Биопсия тканей. Ткань почек, полученную при биопсии, исследуют с помощью электронной микроскопии на наличие ультраструктурных аномалий. Биопсия кожи менее инвазивна, и эксперты рекомендуют выполнять ее в первую очередь.
Генетический анализ. В диагностике синдрома Альпорта, если остаются сомнения после биопсии почки, генетический анализ используется для получения однозначного ответа. Определяются мутации генов синтеза коллагена типа IV.
Аудиометрия. Все дети с семейной историей, позволяющей заподозрить синдром Альпорта, должны проходить высокочастотную аудиометрию для подтверждения нейросенсорной тугоухости. Рекомендуется периодический мониторинг.
Обследование глаз. Обследование у офтальмолога очень важно для раннего выявления и мониторинга переднего лентиконуса и других аномалий.
УЗИ почек. На поздних стадиях синдрома Альпорта ультразвуковое исследование почек помогает выявить структурные нарушения.
Лечение синдрома Альпорта
Синдром Альпорта пока неизлечим. Исследования показали, что ингибиторы АПФ могут уменьшать протеинурию и замедлять прогрессирование почечной недостаточности. Таким образом, использование ИАПФ целесообразно у пациентов с протеинурией, независимо от наличия гипертензии. То же самое касается антагонистов АТII-рецепторов. Оба класса препаратов помогают уменьшить протеинурию путем снижения внутриклубочкового давления. Более того, ингибирование ангиотензина-II, ростового фактора, отвечающего за гломерулярный склероз, теоретически может замедлять склерозирование. Некоторые исследователи предполагают, что циклоспорин способен уменьшать протеинурию и стабилизировать почечную функцию у пациентов с синдромом Альпорта (исследования были небольшими). Но отчеты говорят, что ответ пациентов на циклоспорин очень вариабельный, и иногда препарат может ускорять интерстициальный фиброз.
При почечной недостаточности стандартная терапия включает эритропоэтин для лечения хронической анемии, препараты для контроля остеодистрофии, коррекцию ацидоза и антигипертензивную терапию для контроля АД. Применяется гемодиализ и перитонеальный диализ.
Больным с синдромом Альпорта трансплантация почки не противопоказана: опыт пересадки в США показал хорошие результаты. Генная терапия при разных формах синдрома Альпорта является перспективным вариантом лечения, который сегодня активно изучается западными медицинскими лабораториями.
Вторичная профилактика .
В семьях с Х-сцепленной формой СА при известных мутациях возможна пренатальная диагностика, что крайне важно, если плод мужского пола
– наследственное заболевание почек, вызванное изменением синтеза коллагена типа IV, образующего базальные мембраны почечных клубочков, структуры внутреннего уха, хрусталика глаза. Мужчины страдают развернутой формой болезни с тяжелой симптоматикой. Женщины часто являются носителями гена, оставаясь здоровыми, или проявления болезни у них выражены слабо. Основные симптомы – микрогематурия, протеинурия, почечная недостаточность, сенсорная тугоухость, деформация и вывих хрусталика, катаракта. Диагноз устанавливается согласно клинико-анамнестическим данным, результатам общего анализа мочи, исследования биоптата почки, аудиометрии и офтальмологического осмотра. Лечение симптоматическое, включает терапию иАПФ и БРА.
МКБ-10
Q87.8 Другие уточненные синдромы врожденных аномалий, не классифицированные в других рубриках

Общие сведения
Семейные случаи гематурической нефропатии впервые привлекли внимание исследователей в 1902 году. Спустя почти 30 лет, в 1927 году американский врач А. Альпорт обнаружил частую сочетаемость гематурии с тугоухостью и уремией у мужчин, в то время как у женщин симптомы отсутствовали или были слабовыраженными. Он предположил наследственный характер болезни, которая впоследствии была названа синдромом Альпорта. Синонимы – , гематурический нефрит, семейный гломерулонефрит. Распространенность невысока – 1 случай на 5 тысяч человек. На долю патологии приходится 1% больных с почечной недостаточностью, 2,3% пациентов, перенесших трансплантацию почек. Заболевание диагностируется у людей всех рас, но соотношение различных форм неодинаково.
Причины
По своей природе синдром является гетерогенным наследственным заболеванием – его развитие провоцируется дефектом генов, которые кодируют структуру различных цепей IV типа коллагена. Генетические изменения представлены делециями, сплайсинг, миссенс и нонсенс-мутациями. Их локализация определяет тип наследования болезни:
- X -сцепленный доминантный. Связан с мутацией в локусе COL4A5, который находится на половой хромосоме X. Ген кодирует а5-цепь коллагена 4 типа. Данный генетический дефект обуславливает 80-85% случаев наследственного нефрита. В полной мере заболевание проявляется у мальчиков и мужчин, у представительниц женского пола оставшийся нормальный ген в X-хромосоме компенсирует производство функционального коллагена.
- Аутосомно-рецессивный. Развивается на основе мутаций в генах C0L4A3 и COL4A4. Они локализованы на второй хромосоме, отвечают за структуру а3- и а4-цепи коллагена. Пациенты с этим вариантом синдрома составляют около 15% больных. Выраженность симптомов не зависит от пола.
- Аутосомно-доминантный. Нефрит возникает в результате мутаций генов COL4A3-COLA4, находящихся на 2 хромосоме. Как и в случае аутосомно-рецессивной формой болезни, нарушается синтез а4- и а3-цепей коллагена четвертого типа. Распространенность – 1% всех случаев генетического нефрита.
Патогенез
Гломерулярная базальная мембрана имеет сложное строение, ее образует строгая геометрическая последовательность молекул коллагена 4-го типа и полисахаридные компоненты. При синдроме Альпорта имеются мутации, которые задают дефектное строение спиралевидных коллагеновых молекул. На первых этапах болезни базальная мембрана истончается, начинает расщепляться и расслаиваться. Одновременно возникают утолщенные участки с неравномерными просветлениями. Внутри скапливается тонкогранулярное вещество. Прогрессирование болезни сопровождается полным разрушением базальной гломерулярной мембраны клубочковых капилляров, канальцев почек, структур внутреннего уха и глаз. Таким образом, патогенетически синдром Альпорта представлен четырьмя звеньями: мутацией гена, дефектом строения коллагена, деструкцией базальных мембран, патологией почек (иногда – нарушением слуха и зрения).
Симптомы
Самым распространенным проявлением синдрома Альпорта является гематурия. Микроскопически этот симптом определяется у 95% женщин и у 100% мужчин. При рутинном обследовании мальчиков гематурия обнаруживается уже в первые годы жизни. Другой распространенный признак заболевания – протеинурия. Выведение белка с мочой у пациентов мужского пола с X-сцепленным синдромом начинается в раннем детском возрасте, у остальных – позже. У девочек и женщин уровень экскреции белка повышается незначительно, случаи выраженной протеинурии крайне редки. У всех больных отмечается неуклонное прогрессирование симптома.
Часто у больных формируется нейросенсорная тугоухость . Нарушения слуха дебютируют в детстве, но становятся заметными в подростничестве или молодости. У детей тугоухость распространяется только на звуки высокой частоты, обнаруживается в специально созданных условиях – при аудиометрии. По мере взросления и прогрессирования синдрома нарушается слуховое восприятие средних и низких частот, в том числе человеческой речи. При X-связанном синдроме расстройство слуха к 25 годам имеется у 50% больных мужчин, к 40 годам – у 90%. Тяжесть тугоухости вариабельна, от изменений только в результатах аудиограммы до полной глухоты. Патологии вестибулярного аппарата отсутствуют.
Расстройства зрения включают передний лентиконус – выпячивание центра хрусталика глаза вперед и ретинопатию . Обе патологии проявляются прогрессирующим ухудшением зрительной функции, покраснением, болью в глазах. У некоторых больных имеются стигмы дизэмбриогенеза – анатомические аномалии мочевыделительной системы, глаз, ушных раковин, конечностей. Может наблюдаться высокое расположение неба, укорочение и искривление мизинцев, сращивание пальцев ног, широко расставленные глаза.
Осложнения
Отсутствие лечения больных синдромом Альпорта приводит к быстрому прогрессированию глухоты и слепоты, формированию катаракт . У части пациентов развивается полиневропатия – поражение нервов, сопровождающееся мышечной слабостью, болями, судорогами, тремором, парестезиями, снижением чувствительности. Другим осложнением является тромбоцитопения с высоким риском кровотечений. Наиболее опасным состоянием при наследственном нефрите считается терминальная стадия почечной недостаточности. Больше всего ей подвержены мужчины с типом наследования, сцепленным с половой X-хромосомой. К 60 годам 100% больных этой группы нуждаются в процедурах гемодиализа, перитонеального диализа, трансплантации донорской почки.
Диагностика
В диагностическом процессе принимают участие врачи-нефрологи , урологи, терапевты и генетики. При опросе выясняется возраст дебюта симптомов, наличие у родственников первой линии гематурии, протеинурии или смертельных исходов вследствие ХПН. Для синдрома Альпорта характерно раннее начало и отягощенный семейный анамнез. Дифференциальная диагностика направлена на исключение гематурической формы гломерулонефритов , вторичных нефропатий. Для подтверждения диагноза проводятся следующие процедуры:
- Физикальное обследование. Определяется бледность кожных покров и слизистых оболочек, сниженный мышечный тонус, внешние и соматические признаки дизэмбриогенеза – высокое небо, аномалии строения конечностей, увеличенное расстояние между глазами, сосками. На ранних стадиях болезни диагностируется артериальная гипотония , на поздних – артериальная гипертония.
- Общий анализ мочи. Обнаруживаются эритроциты и повышенное содержание белка – признаки гематурии и протеинурии. Показатель белка мочи напрямую коррелирует с тяжестью синдрома, по его изменению оценивается прогрессирование патологии, вероятность нефротического синдрома , ХПН. Возможно наличие признаков лейкоцитурии абактериального характера.
- Исследование биоптата почек . При микроскопии визуализируется истонченная базальная мембрана, расщепление и разделение ее слоев. На поздней стадии отмечаются утолщенные дистрофичные участки с «сотами» просветления, зоны полной деструкции слоя.
- Молекулярно-генетическое исследование. Генетическая диагностика не является обязательной, но позволяет составить более точный прогноз, подобрать оптимальную схему лечения. Изучается строение генов, мутации в которых обуславливают развитие синдрома. У большей части больных выявляются мутации гена COL4A5.
- Аудиометрия , офтальмологическое исследование . Дополнительно пациентам могут быть назначены диагностические консультации сурдолога и офтальмолога. При аудиометрии обнаруживается снижение слуха: в детском и подростковом возрасте – билатеральная высокочастотная тугоухость, во взрослом возрасте – низкочастотная и среднечастотная тугоухость. Офтальмолог определяет искажение формы хрусталика, поражение сетчатки, наличие катаракты, снижение зрения.
Лечение синдрома Альпорта
Специфическая терапия отсутствует. С раннего возраста проводится активное симптоматическое лечение, снижающее протеинурию. Оно позволяет предотвратить поражение и атрофию почечных канальцев, развитие интерстициального фиброза. С помощью ингибиторов ангиотензинпревращающего фермента и блокаторов рецепторов к ангиотензину II удается приостановить прогрессирование заболевания, добиться регрессии гломерулосклероза, тубулоинтерстициальных и сосудистых изменений в почках. Пациентам с терминальной стадией ХПН назначается гемодиализ , перитонеальный диализ , решается вопрос о целесообразности трансплантации почек .
Прогноз и профилактика
Синдром прогностически благоприятен в случаях, когда гематурия протекает без протеинурии, нет расстройств зрения и тугоухости. Кроме этого, прогноз хороший у большинства женщин – даже при наличии гематурии болезнь прогрессирует медленно, не ухудшает общего состояния. Ввиду наследственного характера патологии предупредить ее развитие невозможно. В семьях, где установлено наличие X-сцепленной формы синдрома, возможно проведение пренатальной диагностики. Генетический скрининг особенно рекомендован женщинам, вынашивающим мальчиков.
Генетическая основа болезни — мутация в гене а-5 цепи коллагена IV типа. Этот тип универсален для базальных мембран почки, кохлеарного аппарата, капсулы хрусталика, сетчатки и роговицы глаза, что доказано в исследованиях с использованием моноклональных антител против этой фракции коллагена. В последнее время указывают на возможность применения ДНК-зондов для пренатальной диагностики наследственного нефрита.
Подчеркивается важность тестирования всех членов семьи с помощью ДНК-зондов для выявления носителей мутантного гена, что имеет большое значение при проведении медико-генетического консультирования семей с этим заболеванием. Однако до 20% семей не имеют родственников, страдающих болезнью почек, что позволяет предполагать высокую частоту спонтанных мутаций аномального гена. У большинства больных наследственным нефритом в семьях имеются лица с почечными заболеваниями, снижением слуха и патологией зрения; имеют значение родственные браки между людьми, имеющими одного или более предков, так как в браке родственных особей возрастает вероятность получения одинаковых генов со стороны обоих родителей. Установлены аутосомно-доминантный и аутосомно-рецессивный и доминантный, сцепленный с Х-хромосомой пути передачи.
У детей чаще различают три варианта наследственного нефрита: синдром Альпорта, наследственный нефрит без тугоухости и семейная доброкачественная гематурия.
Синдром Альпорта — наследственный нефрит с поражением слуха. В основе лежит сочетанный дефект структуры колагена базальной мембраны клубочков почек, структур уха и глаза. Ген классического синдрома Альпорта расположен в локусе 21-22 q длинного плеча Х-хромосомы. В большинстве случаев наследуется по доминантному типу, сцепленному с Х-хромосомой. В связи с этим у мужчин синдром Альпорта протекает тяжелее, так как у женщин функция мутантного гена компенсируется здоровым аллелем второй, неповрежденной хромосомы.
Генетической основой развития наследственного нефрита являются мутации в генах альфа-цепей коллагена IV типа. Известно шесть а-цепей IV коллагена Г типа: гены а5- и а6-цепей (Соl4A5 и Соl4A5) находятся на длинном плече Х-хромосомы в зоне 21-22q; гены а3- и а4-цепей (Соl4A3 и Соl4A4) — на 2-й хромосоме; гены a1- и a2-цепей (Соl4A1 и Соl4A2) — на 13-й хромосоме.
В большинстве случаев (80-85%) выявляется Х-сцепленный тип наследования заболевания, связанный с повреждениями гена Соl4A5 в результате делеции, точечных мутаций или нарушений сплайсинга. В настоящее время найдено более 200 мутаций гена Соl4A5, ответственных за нарушение синтеза а5-цепей коллагена IV типа. При этом типе наследования заболевание проявляется у детей обоего пола, но у мальчиков протекает тяжелее.
Мутации в локусах генов Соl4A3 и Соl4A4, ответственных за синтез а3- и а4 — цепей коллагена IV типа, наследуются аутосомно. По данным исследований аутосомно-доминантныи тип наследования отмечается в 16% случаев наследственного нефрита, аутосомно-рецессивный — у 6% больных. Изестно около 10 вариантов мутаций генов Соl4A3 и Соl4A4.
Результатом мутаций является нарушение процессов сборки коллагена IV типа, приводящее к нарушению его структуры. Коллаген IV типа является одним из основных компонентов гломерулярной базальной мембраны, кохлеарного аппарата и хрусталика глаза, патология которых будет выявляться в клинике наследственного нефрита.
Коллаген IV типа, входящий в состав гломерулярной базальной мембраны, состоит в основном из двух а1-цепей (IV) и одной а2-цепи (IV), а также содержит а3, а4, а5-цепи. Наиболее часто при Х-сцепленном наследовании мутация гена Соl4A5 сопровождается отсутствием а3-, а4-, а5- и а6 цепей в структуре коллагена IV типа, а количество о1- и а2-цепей в гломерулярной базальной мембране возрастает. Механизм этого феномена неясен, предполагается, что причиной являются посттранскрипционные изменения мРНК.
Отсутствие а3-, а4- и а5-цепей в структуре IV типа коллагена базальных мембран клубочков приводит к их истончению и ломкости на ранних стадиях синдрома Альпорта, что клинически проявляется чаще гематурией (реже гематурией с протеинурией или только протеинурией), снижением слуха и лентиконусом. Дальнейшее прогрессирование заболевания приводит к утолщению и нарушению проницаемости базальных мембран на поздних стадиях заболевания, с разрастанием в них коллагена V и VI типов, проявляющихся в нарастании протеинурии и снижении почечных функций.
Характер мутации, лежащей в основе наследственного нефрита, во многом определяет его фенотипическое проявление. При делеции Х-хромосомы с одновременной мутацией генов Соl4A5 и Соl4A6, ответственных за синтез а5- и а6-цепей коллагена IV типа, синдром Альпорта сочетается с лейомиоматозом пищевода и половых органов. По данным исследований при мутации гена Соl4A5, связанной с делецией, отмечаются большая тяжесть патологического процесса, сочетание почечного поражения с экстраренальными проявлениями и ранним развитием хронической почечной недостаточности, по сравнению сточечной мутацией этого гена.
Морфологически при электронной микроскопии выявляется истончение и расслоение гломерулярных базальных мембран (особенно lamina densa) и наличие электронно-плотных гранул. Поражение гломерул может быть неоднородным у одного и того же больного, от минимального фокального поражения мезангия до гломерулосклероза. Гломерулит при синдроме Альпорта всегда носит иммунонегативный характер, что отличает его от гломерулонефрита. Характерны развитие атрофии канальцев, лимфогистиоцитарная инфильтрация, наличие «пенистых клеток» с включениями липидов — липофагами. При прогрессировании заболевания выявляется утолщение и выраженная деструкция базальных мембран клубочков.
Выявляются определенные сдвиги в состоянии имунной системы. У больных наследственным нефритом отмечено снижение уровня Ig A и склонность к повышению концентрации IgМ в крови, уровень IgG может быть повышен на ранних стадиях развития заболевания и снижаться на поздних сроках. Возможно, повышение концентрации IgM и G является своеобразной компенсаторной реакцией в ответ на дефицит IgA.
Функциональная активность системы Т-лимфоцитов снижена; отмечается избирательное снижение В-лимфоцитов, ответственных за синтез Ig A, нарушается фагоцитарное звено иммунитета, в основном за счет нарушения процессов хемотаксиса и внутриклеточного переваривания в нейтрофилах
При исследовании биоптата почек у больных с синдромом Альпорта по данным электронной микроскопии наблюдаются ультраструктурные изменения базальной мембраны клубочка: истончение, нарушение структуры и расщепление гломерулярных базальных мембран с изменением ее толщины и неравномерностью контуров. На ранних стадиях наследственного нефрита дефект определяет истончение и ломкость гломерулярных базальных мембран.
Истончение гломерулярных мембран является более благоприятным признаком и чаще встречается у девочек. Более постоянный электронно-микроскопический признак при наследственном нефрите — расщепление базальной мембраны, причем выраженность деструкции ее коррелирует с тяжестью процесса.
Причины и механизм развития синдрома Альпорта
Синдром Альпорта вызван мутациями в генах COL4A4, COL4A3, COL4A5, отвечающих за биосинтез коллагена. Мутации в указанных генах нарушают нормальный синтез коллагена типа IV, который является очень важным структурным компонентом базальных мембран в почках, внутреннем ухе и глазах.
Базальные мембраны – это тонкие пленочные структуры, которые поддерживают ткани и отделяют их друг от друга. При нарушении синтеза коллагена типа IV гломерулярные базальные мембраны в почках не способны нормально фильтровать токсичные продукты из крови, пропуская в мочу белки (протеинурия) и эритроциты (гематурия). Аномалии синтеза коллагена типа IV приводят к почечной недостаточности и отказу почек, что и является главной причиной смерти при синдроме Альпорта.
Клиника
Гематурия – это наиболее частое и раннее проявление синдрома Альпорта. Микроскопическая гематурия наблюдается у 95% женщин и практически у всех мужчин. У мальчиков гематурию обычно выявляют в первые годы жизни. Если у мальчика за первые 10 лет жизни не обнаружена гематурия, то американские эксперты рекомендуют считать, что у него маловероятно наличие синдрома Альпорта.
Протеинурия в детстве обычно отсутствует, но иногда развивается у мальчиков с Х-сцепленным синдромом Альпорта. Протеинурия, как правило, прогрессирует. Значительная протеинурия у больных женского пола встречается нечасто.
Гипертензия чаще присутствует у пациентов мужского пола с XLAS и у больных обоих полов с ARAS. Частота и тяжесть гипертензии повышается с возрастом и по мере прогрессирования почечной недостаточности.
Нейросенсорная тугоухость (нарушение слуха) – это характерное проявление синдрома Альпорта, которое наблюдается довольно часто, но не всегда. Есть целые семьи с синдромом Альпорта, которые страдают от тяжелой нефропатии, но имеют нормальный слух. Нарушение слуха никогда не обнаруживается при рождении. Билатеральная высокочастотная нейросенсорная тугоухость обычно проявляется в первые годы жизни или в раннем подростковом возрасте. На ранней стадии болезни нарушение слуха определяется только при аудиометрии.
По мере прогрессирования, нарушение слуха распространяется на низкие частоты, включая человеческую речь. После появления тугоухости следует ожидать вовлечения почек. Американские ученые утверждают, что при Х-связанном синдроме Альпорта 50% мужчин страдают нейросенсорной тугоухостью к 25 годам, а к 40 годам – около 90%.
Передний лентиконус (выпячивание центрального участка хрусталика глаза вперед) наблюдается у 25% пациентов с XLAS. Лентиконуса нет при рождении, но с годами он приводит к прогрессирующему ухудшению зрения, которое заставляет больных часто менять очки. Состояние не сопровождается болью в глазах, покраснением или нарушениями цветового зрения.
Ретинопатия – это самое распространенное проявление синдрома Альпорта со стороны органа зрения, поражает 85% мужчин с Х-сцепленной формой болезни. Появление ретинопатии обычно предшествует почечной недостаточности.
Задняя полиморфная дистрофия роговицы – редкое состояние при синдроме Альпорта. У большинства нет никаких жалоб. Мутация L1649R в гене коллагена COL4A5 может также вызывать истончение сетчатки, которое ассоциируется с Х-сцепленным синдромом Альпорта.
Диффузный лейомиоматоз пищевода и бронхиального дерева – еще одно редкое состояние, которое наблюдается в некоторых семьях с синдромом Альпорта. Симптомы появляются в позднем детском возрасте и включают нарушение глотания (дисфагия), рвоту, боль в эпигастрии и за грудиной, частые бронхиты, одышку, кашель. Лейомиоматоз подтверждается компьютерной томографией или МРТ.
Аутосомно-рецессивная форма синдрома Альпорта
На ARAS приходится всего 10-15% случаев болезни. Эта форма встречается у детей, чьи родители являются носителями одного из пораженных генов, сочетание которых у ребенка вызывает болезнь. Сами родители не имеют симптомов или имеют незначительные проявления, а дети тяжело больны – их симптомы напоминают XLAS.
Аутосомно-доминантная форма синдрома Альпорта
ADAS – это самая редкая форма синдрома, которая затрагивает одно поколение за другим, причем мужчины и женщины болеют одинаково тяжело. Почечные проявления и глухота напоминают XLAS, но почечная недостаточность может возникать в более позднем возрасте. Клинические проявления ADAS дополняются склонностью к кровотечениям, макротромбоцитопенией, синдромом Эпштейна, наличием нейтрофильных включений в крови.
Диагностика синдрома Альпорта
Лабораторные анализы. Анализ мочи: у больных с синдромом Альпорта чаще всего присутствует кровь в моче (гематурия), а также высокое содержание белка (протеинурия). Анализы крови демонстрирует почечную недостаточность.
Биопсия тканей. Ткань почек, полученную при биопсии, исследуют с помощью электронной микроскопии на наличие ультраструктурных аномалий. Биопсия кожи менее инвазивна, и американские эксперты рекомендуют выполнять ее в первую очередь.
Генетический анализ. В диагностике синдрома Альпорта, если остаются сомнения после биопсии почки, генетический анализ используется для получения однозначного ответа. Определяются мутации генов синтеза коллагена типа IV.
Аудиометрия. Все дети с семейной историей, позволяющей заподозрить синдром Альпорта, должны проходить высокочастотную аудиометрию для подтверждения нейросенсорной тугоухости. Рекомендуется периодический мониторинг.
Обследование глаз. Обследование у офтальмолога очень важно для раннего выявления и мониторинга переднего лентиконуса и других аномалий.
УЗИ почек. На поздних стадиях синдрома Альпорта ультразвуковое исследование почек помогает выявить структурные нарушения.
Британские специалисты, основываясь на новых данных (2011) по генетическим мутациям у пациентов с Х-сцепленным синдромом Альпорта, рекомендуют анализ на мутации гена COL4A5, если пациент отвечает хотя бы двум диагностическим критериям по Gregory, и анализ COL4A3 и COL4A4, если мутация COL4A5 не обнаружена или подозревается аутосомное наследование.
Лечение синдрома Альпорта
Синдром Альпорта пока неизлечим. Исследования показали, что ингибиторы АПФ могут уменьшать протеинурию и замедлять прогрессирование почечной недостаточности. Таким образом, использование ИАПФ целесообразно у пациентов с протеинурией, независимо от наличия гипертензии. То же самое касается антагонистов АТII-рецепторов. Оба класса препаратов, судя по всему, помогают уменьшить протеинурию путем снижения внутриклубочкового давления. Более того, ингибирование ангиотензина-II, ростового фактора, отвечающего за гломерулярный склероз, теоретически может замедлять склерозирование.
Некоторые исследователи предполагают, что циклоспорин способен уменьшать протеинурию и стабилизировать почечную функцию у пациентов с синдромом Альпорта (исследования были небольшими). Но отчеты говорят, что ответ пациентов на циклоспорин очень вариабельный, и иногда препарат может ускорять интерстициальный фиброз.
При почечной недостаточности стандартная терапия включает эритропоэтин для лечения хронической анемии, препараты для контроля остеодистрофии, коррекцию ацидоза и антигипертензивную терапию для контроля АД. Применяется гемодиализ и перитонеальный диализ. Больным с синдромом Альпорта трансплантация почки не противопоказана: опыт пересадки в США показал хорошие результаты.
Генная терапия при разных формах синдрома Альпорта является перспективным вариантом лечения, который сегодня активно изучается западными медицинскими лабораториями.
Истинные причины заболевания
Истинные причины синдрома альпорта до сих полностью не изучены учеными. В нашем организме есть ген, функциональной обязанностью которого является обмен белка в тканях почек. Так вот мутация этого гена и является наиболее вероятной причиной появления недуга.
Теперь рассмотрим провоцирующие факторы, которые могут способствовать появлению заболевания. К ним можно отнести:
- тяжелые инфекционные процессы;
- прививки;
- сильные физические нагрузки.
Как видно из многочисленных случаев медицинской практики, порой развитию синдрома альпорта может способствовать обычная острая респираторная вирусная инфекция. Именно ввиду таких высоких рисков заболеваемости дети, которые имеют отягощенную наследственность, должны чаще проходить регулярное диагностическое обследование.
Впервые это заболевание было зарегистрировано в начале прошлого столетия. Врач наблюдал за семьей, в которой в течение нескольких поколений наблюдалась гематурия. Позже была замечена связь между гематурией и тугоухостью, а также поражением глаз. Позже, когда медицина совершенствовалась, медиками глубже исследовали генетическую природу данного синдрома.
В большинстве случаев у ее «обладателей» имеются родственники с почечными патологиями и другими признаками данного синдрома. Роль играют также родственные браки, в результате которых у ребёнка возрастает вероятность получения одинаковых генов. У больных с синдромом Альпорта выявляются сдвиги со стороны иммунной системы.
Симптомы
Наследственный нефрит имеет ярко выраженную клиническую симптоматику. Если говорить о начальных стадиях патологического процесса, то он проявляется следующим образом:
- появление крови в моче;
- ухудшение зрительной функции;
- нарушения слуховой способности, вплоть до развития глухоты.
Клинические симптомы будут нарастать по мере развития заболевания. С течением времени появляются признаки интоксикации, а также развивается анемия. Общее состояние и возраст пациента влияет на выраженность клинической картины.
Другими характерными симптомами заболевания являются такие признаки:
- сильные головные боли;
- мышечные боли;
- даже небольшая физическая активность быстро утомляет пациента;
- головокружение;
- артериальная гипертензия, которая сменяется резким падением давления;
- одышка;
- поверхностное дыхание;
- шум в ушах, который приобретает постоянный характер.
Если же речь идет о хронической форме наследственного нефрита, клиническая картина здесь будет немного отличаться, а именно:
- слабость и общее недомогание;
- частые позывы к мочеиспусканию, примеси крови;
- мочеиспускание не приносит облегчения;
- тошнота и рвота;
- ухудшение аппетита и, как следствие, потеря веса;
- кровоизлияние;
- зуд кожи;
- судороги;
- болезненные ощущения в грудной клетке;
- в тяжелых случаях появляется спутанность сознания и приступы беспамятства.
Инфекционные процессы дыхательных путей, активные физические нагрузки, профилактические прививки – все это может спровоцировать усиление гематурии. Что касается присутствия белка в моче, то сначала протеинурия имеет непостоянный характер, а затем постепенно нарастает и становится стойкой.
Нарастают также симптомы интоксикации, тугоухость особенно наблюдается у мальчиков, дети быстро устают, их беспокоят сильные головные боли. Дети значительно отстают в физическом развитии.
Виды
Специалисты различают три разновидности синдрома альпорта:
- ярко выраженные симптомы и быстрое прогрессирование острой почечной недостаточности;
- быстро прогрессирует заболевание, но при этом не наблюдается нарушение зрения и слуха;
- доброкачественное течение недуга, при котором отсутствуют клинические симптомы и прогрессирование. В этом варианте развития прогноз благоприятен. Если же у женщины присутствует аутосомно-рецессивный тип наследования, тогда наблюдается более тяжелое течение недуга.
Диагностическое обследование
Если присутствуют подозрения на наследственный фактор у детей, следует как можно раньше обратиться к детскому врачу. Для уточнения диагноза применяются лабораторные и инструментальные методы исследования. Что касается лабораторной диагностики, то она включает в себя общий анализ крови и мочи, а также биохимическое исследование.
Если говорить об инструментальной диагностике, то сюда можно отнести следующее:
- ультразвуковое исследование почек и надпочечников;
- биопсию почек;
- рентгенограмму почек.
Иногда могут понадобиться дополнительные генетические анализы. Больным назначается консультация нефролога и дополнительно – генетика.
Основными критериями диагностического обследования являются:
- присутствие в семье двух человек с нефропатией;
- гематурия является доминирующим симптомом;
- тугоухость у кого-то из членов семьи;
- появление хронической почечной недостаточности у кого-нибудь из родственников.
Если говорить о дифференциальном анализе, то наследственный нефрит сравнивают с приобретенной формой гломерулонефрита, при котором наблюдается также гематурия. В чем же отличие? Гломерулонефрит имеет острое начало и существует прямая связь с перенесенной инфекцией. Если наследственный нефрит проявляется в виде артериальной гипотонии, то гломерулонефрит, напротив, выражается в артериальной гипертензии.
Методы борьбы
Лечение синдрома альпорта включает в себя сочетание медикаментозных средств и специального диетического питания. Стоит отметить, что специфических лекарственных средств, которые бы устраняли именно этот генетический недуг, до сих пор не существует. Направленность лекарственных средств, применяемых при наследственном нефрите, связана с нормализацией работы почек. Диетическое питание для детей прописывается врачом индивидуально. Как правило, таких предписаний необходимо придерживаться всю оставшуюся жизнь. Показаны прогулки на свежем воздухе. В крайних случаях специалист может принять решение о проведении оперативного вмешательства. Обычно операция проводится при достижении пятнадцатилетнего возраста.
Правильное питание
Сразу же хочется отметить продукты питания, которые необходимо исключить из рациона. К ним относятся:
- соленая, жирная и копченая пища;
- пряности и острая еда;
- алкогольные напитки, но иногда врачи могут назначать красное вино в лечебных целях;
- продукты, которые имеют в своем составе искусственные красители.
Пища должна быть витаминизированной и калорийной, но при этом без высокого содержания белка. Физические нагрузки исключаются. Занятия спортом, особенно это касается детей, могут проходить только в том случае, если они не запрещены врачом.
Пища должна быть полноценной и в достаточном количестве содержать белки, жиры и углеводы, при этом должны учитываться функциональные способности почек.
Почечная недостаточность является одним из наиболее тяжелых осложнений синдрома альпорта. Согласно статистике, недостаточностью страдают мальчики от шестнадцати до двадцати лет. Если будет отсутствовать адекватное лечение и правильный способ жизни, то летальный исход наступает раньше, чем в тридцать лет.
Кроме этого, важно избегать контакта с инфекционными больными для снижения рисков развития респираторных заболеваний. Профилактические прививки противопоказаны детям с наследственным нефритом, а вакцинация может проводиться лишь по эпидемиологическим показаниям.
Профилактики синдрома альпорта нет. Это генетический недуг, который предупредить невозможно. Если же у ребенка был диагностирован недуг, тогда следует придерживаться рекомендаций врача и правильного образа жизни.
Если говорить о прогнозах, то крайне неблагоприятными являются следующие критерии:
- мужской пол;
- наличие хронической почечной недостаточности у членов семьи;
- неврит слухового нерва;
- наличие белка в моче.
Больным назначаются лекарственные средства, которые влияют на улучшение обмена веществ:
- витамин А, Е;
- пиридоксин;
- кокарбоксилаза.
Трансплантация – это наиболее эффективный метод лечения при синдроме альпорта. Рецидив заболевания в трансплантате не отмечается, и лишь в незначительных случаях возможно развитие нефрита.
Итак, синдром альпорта – это серьезный недуг, требующий своевременного и грамотного подхода к лечению. Не существует профилактики наследственного нефрита, но его течение можно облегчить при неукоснительном соблюдении всех врачебных рекомендаций.